rgbio provides performant reading and writing operations for GenBank (.gb/.gbk/.gbff) files in R via an interface to the high-performance gb-io Rust crate. It is designed to be fast and memory-efficient while providing R-friendly data structures.
Why rgbio?
- the only way to directly write GenBank files from R (to my knowledge)
- much faster reading of GenBank files (~15x-25x faster than other packages in my benchmarks)
- reading into and writing from both: tidy objects (e.g. tibbles/data.frames) and “Bioconductor Sequence Infrastructure” objects (e.g. DNAStrings).
- robust parsing via the robust
gb-ioRust crate - extensively tested on ~50 diverse GenBank files with many edge cases.
Installation
The rgbio package is not available on CRAN (for now), because it depends on a Rust crate. You can install it from the R-universe repository without having installed Rust or any Rust toolchain, as there are binary versions available for Windows, macOS, and Linux.
install.packages("rgbio",
repos = c("https://richardstoeckl.r-universe.dev",
"https://cloud.r-project.org"))If there is no pre-built binary available for your system, or you want the current development snapshot, you can install rgbio from GitHub, provided you have the Rust toolchain installed. You can find information on how to install Rust at https://github.com/r-rust/hellorust.
# install.packages("remotes")
remotes::install_github("richardstoeckl/rgbio")Usage
Reading GenBank files
Reading files is as simple as providing the path (and optionally chosing the output type):
Writing GenBank files
Minimum required inputs:
-
file: output file path -
sequences: non-empty named character vector orDNAStringSet
Optional inputs:
features = NULLmetadata = NULLappend = FALSEvalidate = TRUEline_width = 80
If metadata is omitted, rgbio fills defaults per record:
-
name,definition,accessionfrom the sequence name molecule_type = "DNA"
append = TRUE requires that the target file already exists and is a valid GenBank file.
Minimal Bioconductor Example
library(rgbio)
out <- tempfile(fileext = ".gb")
seqs <- Biostrings::DNAStringSet(c(min2 = "ATGCGG"))
write_gbk(
file = out,
sequences = seqs
)Performance
Performance comparison reading 17 real-world GenBank files (rgbio v0.3.0):
| Parser | Relative Speed | Median Time (ms) |
|---|---|---|
| rgbio::read_gbk(format = ‘tidy’) | 24.6x | 64.5 |
| rgbio::read_gbk(format = ‘bioconductor’) | 22.4x | 70.8 |
| geneviewer::read_gbk() | 1.3x | 1193.6 |
| genbankr::readGenBank() | baseline | 1583.6 |
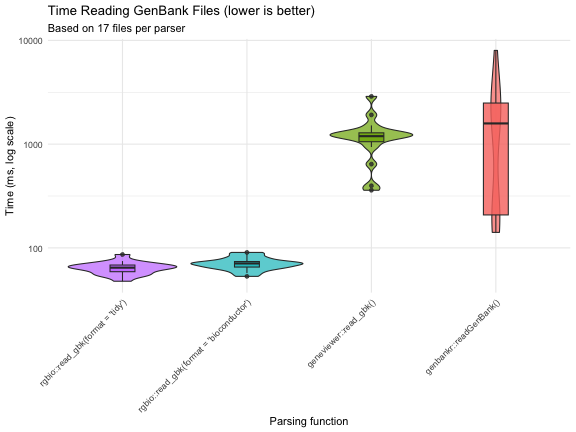
Full benchmark details and methodology available in the benchmarks article.
Disclaimer
Important note: This was/is a project for me to play around with agentic coding, and was written primarily by LLMs (“AI”) under my direction. Nevertheless, it provides real value as it is one of the only ways to write GenBank files in R, and is one of the most performant ways to read Genbank files to R. It uses the very robust Rust gb-io crate and is tested against ~50 diverse GenBank files with many edge cases.