Benchmarking rgbio Performance
benchmarks.RmdBenchmarking rgbio Performance
Benchmark Setup
To evaluate real-world performance, we downloaded 17 archaeal type
strain assemblies from the order “Methanococcales” directly from NCBI
using the datasets command-line tool. These represent
real-world GenBank files with varying sizes and complexity.
#> The Benchmark was run on 2026-05-27 using rgbio version 0.4.0Data Download Command
# Download 17 archaeal type strain assemblies in GenBank format
datasets download genome taxon "methanococcales" \
--from-type \
--reference \
--assembly-source RefSeq \
--include gbff \
--filename methanococcales_type_strains.zip --dehydrated && \
unzip methanococcales_type_strains.zip && \
datasets rehydrate --directory .Packages Tested
We compared rgbio against three other popular GenBank
reading packages:
library(bench)
library(dplyr)
library(ggplot2)
# Packages being benchmarked
library(rgbio) # High-performance GenBank I/O using Rust gb-io crate
library(read.gb) # Specifically made for reading gb files
library(geneviewer) # Has a function to read gb files
library(genbankr) # Traditional Bioconductor GenBank reader. Only available up to Bioconductor version: 3.16 ≙ R version 4.2-
rgbio: Our Rust-powered package with two output
formats (
tidyandbioconductor) - geneviewer: A visualization-focused package with GenBank reading capabilities
- genbankr: The traditional Bioconductor solution for GenBank parsing
- read.gb: A dedicated GenBank file reader
Running the Benchmarks
# Set up test data paths
gbk_dir <- "./ncbi_dataset/data"
gbk_files <- list.files(gbk_dir, pattern = "\\.gbff$", recursive = TRUE, full.names = TRUE)
# Define reader functions for consistent testing
readers <- list(
"rgbio::read_gbk(format = 'tidy')" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "tidy")
}, error = function(e) {
message("rgbio (tidy) failed on ", file, ": ", e$message)
return(NULL)
})
},
"rgbio::read_gbk(format = 'bioconductor')" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "bioconductor")
}, error = function(e) {
message("rgbio (bioconductor) failed on ", file, ": ", e$message)
return(NULL)
})
},
"rgbio::read_gbk(format = 'base')" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "base")
}, error = function(e) {
message("rgbio (base) failed on ", file, ": ", e$message)
return(NULL)
})
},
"rgbio::read_gbk(format = 'tidy', validate = FALSE)" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "tidy", validate = FALSE)
}, error = function(e) {
message("rgbio (tidy) failed on ", file, ": ", e$message)
return(NULL)
})
},
"rgbio::read_gbk(format = 'bioconductor', validate = FALSE)" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "bioconductor", validate = FALSE)
}, error = function(e) {
message("rgbio (bioconductor) failed on ", file, ": ", e$message)
return(NULL)
})
},
"rgbio::read_gbk(format = 'base', validate = FALSE)" = function(file) {
tryCatch({
rgbio::read_gbk(file, format = "base", validate = FALSE)
}, error = function(e) {
message("rgbio (base) failed on ", file, ": ", e$message)
return(NULL)
})
},
"geneviewer::read_gbk()" = function(file) {
tryCatch({
geneviewer::read_gbk(file)
}, error = function(e) {
message("geneviewer failed on ", file, ": ", e$message)
return(NULL)
})
},
"genbankr::readGenBank()" = function(file) {
tryCatch({
genbankr::readGenBank(file)
}, error = function(e) {
message("genbankr failed on ", file, ": ", e$message)
return(NULL)
})
}
)We use the bench package to get accurate timing
measurements, testing each parser on every file exactly once to simulate
real-world usage patterns.
# Run comprehensive benchmarks across all file × reader combinations
benchmark_results <- press(
file = gbk_files,
reader = names(readers),
{
result <- bench::mark(
result = readers[[reader]](file),
iterations = 1,
check = FALSE
)
}
)To benchmark the writing, we pre-read each file once per
rgbio format (without benchmarking), store those parsed
objects, and then benchmark only the write step.
# Pre-read objects once (no benchmarking) for each file × rgbio format
write_cache_grid <- expand.grid(
file = gbk_files,
write_format = c("tidy", "bioconductor"),
stringsAsFactors = FALSE
)
write_cache_read_once <- function(file, write_format) {
tryCatch({
rgbio::read_gbk(file, format = write_format)
}, error = function(e) {
message("rgbio ", write_format, " pre-read failed on ", file, ": ", e$message)
return(NULL)
})
}
rgbio_cached_reads <- write_cache_grid %>%
rowwise() %>%
mutate(parsed_obj = list(write_cache_read_once(file, write_format))) %>%
ungroup()
# Benchmark write phase only, reusing cached parsed objects
write_benchmark_results <- press(
row_id = seq_len(nrow(rgbio_cached_reads)),
{
file <- rgbio_cached_reads$file[[row_id]]
write_format <- rgbio_cached_reads$write_format[[row_id]]
obj <- rgbio_cached_reads$parsed_obj[[row_id]]
result <- bench::mark(
result = {
tmp_file <- tempfile(pattern = "rgbio_write_benchmark_", fileext = ".gb")
tryCatch({
if (is.null(obj)) {
stop("cached read object is NULL")
}
if (write_format == "tidy") {
seqs <- setNames(as.character(obj$sequences$sequence), obj$sequences$record_id)
rgbio::write_gbk(
file = tmp_file,
sequences = seqs,
features = obj$features,
metadata = obj$metadata
)
} else {
rgbio::write_gbk(
file = tmp_file,
sequences = obj$sequences,
features = obj$features,
metadata = obj$metadata
)
}
}, error = function(e) {
message("rgbio ", write_format, " write failed on ", file, ": ", e$message)
return(FALSE)
}, finally = {
unlink(tmp_file)
})
},
iterations = 1,
check = FALSE
) %>%
mutate(
file = file,
write_format = write_format,
operation = "write"
)
}
)
# Store write benchmark results for downstream processing
write_results <- write_benchmark_resultsProcessing Results
# Clean up results and add useful metadata
benchmark_df <- benchmark_results %>%
mutate(
file_id = as.numeric(factor(file)),
file_name = dirname(file),
file_size_mb = file.info(file)$size / (1024^2),
success = !is.na(median),
time_ms = ifelse(success, as.numeric(median) * 1000, NA),
memory_mb = ifelse(success, as.numeric(gsub("[^0-9.]", "", mem_alloc)), NA)
) %>%
select(reader, file_id, file_name, file_size_mb, success, time_ms, memory_mb)
# Parse write benchmark results with matching metadata fields
write_benchmark_df <- write_results %>%
mutate(
file_id = as.numeric(factor(file)),
file_name = dirname(file),
file_size_mb = file.info(file)$size / (1024^2),
success = !is.na(median),
time_ms = ifelse(success, as.numeric(median) * 1000, NA),
memory_mb = ifelse(success, as.numeric(gsub("[^0-9.]", "", mem_alloc)), NA)
) %>%
select(write_format, operation, file_id, file_name, file_size_mb, success, time_ms, memory_mb)Results
Performance Visualization
p1 <- ggplot(benchmark_df, aes(x = factor(reader, levels = names(readers)), y = time_ms, fill = reader)) +
geom_violin(alpha = 0.7) +
geom_boxplot(width = 0.2, alpha = 0.8) +
scale_y_log10() +
labs(
title = "Time Reading GenBank Files (lower is better)",
y = "Time (ms, log scale)",
subtitle = paste("Based on",
benchmark_df %>%
filter(success) %>%
group_by(reader) %>%
summarise(n = n()) %>%
pull(n) %>%
min(),
"files per parser")
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
xlab("Parsing function")
p1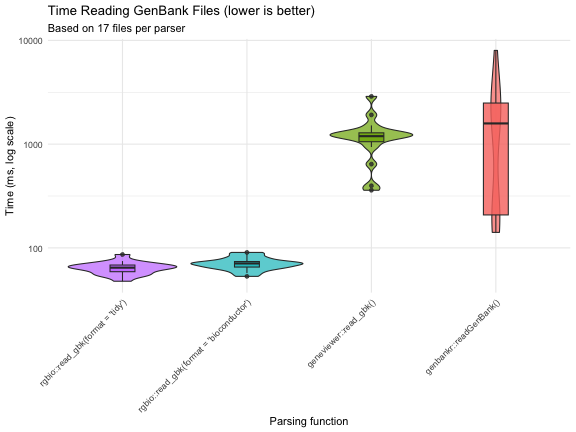
Summary Statistics
# Calculate performance summary with speedup factors
summary_stats <- benchmark_df %>%
group_by(reader) %>%
summarise(
n_files = n(),
median_time_ms = round(median(time_ms, na.rm = TRUE), 1),
mean_time_ms = round(mean(time_ms, na.rm = TRUE), 1),
median_memory_mb = round(median(memory_mb, na.rm = TRUE), 1),
.groups = "drop"
) %>%
arrange(factor(reader, levels = names(readers))) %>%
mutate(
max_time = max(median_time_ms),
speedup = case_when(
median_time_ms == max_time ~ "baseline",
TRUE ~ paste0(round(max_time / median_time_ms, 1), "x")
)
) %>%
select(reader, speedup, median_time_ms, mean_time_ms, median_memory_mb, n_files)
knitr::kable(summary_stats,
col.names = c("Parser", "Speed vs Slowest", "Median Time (ms)",
"Mean Time (ms)", "Median Memory (MB)", "Files Tested"),
caption = "Performance comparison summary")| Parser | Speed vs Slowest | Median Time (ms) | Mean Time (ms) | Median Memory (MB) | Files Tested |
|---|---|---|---|---|---|
| rgbio::read_gbk(format = ‘tidy’) | 45.3x | 42.2 | 42.5 | 4.4 | 17 |
| rgbio::read_gbk(format = ‘bioconductor’) | 38.2x | 50.1 | 48.8 | 6.2 | 17 |
| rgbio::read_gbk(format = ‘base’) | 45.4x | 42.1 | 41.6 | 4.3 | 17 |
| rgbio::read_gbk(format = ‘tidy’, validate = FALSE) | 44.8x | 42.7 | 44.2 | 5.0 | 17 |
| rgbio::read_gbk(format = ‘bioconductor’, validate = FALSE) | 37.2x | 51.4 | 68.3 | 5.0 | 17 |
| rgbio::read_gbk(format = ‘base’, validate = FALSE) | 45.6x | 41.9 | 42.5 | 4.1 | 17 |
| geneviewer::read_gbk() | 1.5x | 1261.0 | 1387.9 | 1.5 | 17 |
| genbankr::readGenBank() | baseline | 1912.1 | 1688.6 | 38.7 | 17 |
Key Findings
The benchmarks reveal several important insights:
-
rgbio is significantly faster: Both
rgbioformats substantially outperform traditional R-based parsers -
Consistent performance:
rgbioshows reliable performance across different file sizes and complexities
-
Format flexibility: The
tidyformat offers the best performance, whilebioconductorformat provides compatibility with existing workflows -
Memory efficiency:
rgbiomaintains competitive memory usage while delivering superior speed
Technical Details
sessionInfo()
#> R version 4.2.3 (2023-03-15)
#> Platform: aarch64-apple-darwin20 (64-bit)
#> Running under: macOS 15.7.7
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] C/UTF-8/C/C/C/C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] genbankr_1.26.0 geneviewer_0.1.11 read.gb_2.2 rgbio_0.4.0
#> [5] ggplot2_4.0.3 dplyr_1.2.1 bench_1.1.4
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-9 matrixStats_1.5.0
#> [3] fontawesome_0.5.3 bit64_4.8.2
#> [5] filelock_1.0.3 RColorBrewer_1.1-3
#> [7] progress_1.2.3 httr_1.4.8
#> [9] GenomeInfoDb_1.34.9 tools_4.2.3
#> [11] utf8_1.2.6 R6_2.6.1
#> [13] otel_0.2.0 DBI_1.3.0
#> [15] BiocGenerics_0.44.0 withr_3.0.2
#> [17] tidyselect_1.2.1 prettyunits_1.2.0
#> [19] bit_4.6.0 curl_7.1.0
#> [21] compiler_4.2.3 cli_3.6.6
#> [23] Biobase_2.58.0 xml2_1.5.2
#> [25] DelayedArray_0.24.0 rtracklayer_1.58.0
#> [27] scales_1.4.0 S7_0.2.2
#> [29] rappdirs_0.3.4 stringr_1.6.0
#> [31] digest_0.6.39 Rsamtools_2.14.0
#> [33] rentrez_1.2.4 XVector_0.38.0
#> [35] pkgconfig_2.0.3 htmltools_0.5.9
#> [37] MatrixGenerics_1.10.0 dbplyr_2.5.2
#> [39] fastmap_1.2.0 BSgenome_1.66.3
#> [41] htmlwidgets_1.6.4 rlang_1.2.0
#> [43] RSQLite_3.52.0 BiocIO_1.8.0
#> [45] farver_2.1.2 generics_0.1.4
#> [47] jsonlite_2.0.0 BiocParallel_1.32.6
#> [49] VariantAnnotation_1.44.1 RCurl_1.98-1.18
#> [51] magrittr_2.0.5 GenomeInfoDbData_1.2.9
#> [53] Matrix_1.5-3 Rcpp_1.1.1-1.1
#> [55] S4Vectors_0.36.2 lifecycle_1.0.5
#> [57] stringi_1.8.7 yaml_2.3.12
#> [59] SummarizedExperiment_1.28.0 zlibbioc_1.44.0
#> [61] BiocFileCache_2.6.1 grid_4.2.3
#> [63] blob_1.3.0 parallel_4.2.3
#> [65] crayon_1.5.3 profmem_0.7.0
#> [67] lattice_0.22-9 Biostrings_2.66.0
#> [69] GenomicFeatures_1.50.4 hms_1.1.4
#> [71] KEGGREST_1.38.0 knitr_1.51
#> [73] pillar_1.11.1 GenomicRanges_1.50.2
#> [75] rjson_0.2.23 codetools_0.2-20
#> [77] biomaRt_2.54.1 stats4_4.2.3
#> [79] XML_3.99-0.23 glue_1.8.1
#> [81] evaluate_1.0.5 png_0.1-9
#> [83] vctrs_0.7.3 gtable_0.3.6
#> [85] purrr_1.2.2 tidyr_1.3.2
#> [87] cachem_1.1.0 xfun_0.57
#> [89] restfulr_0.0.16 tibble_3.3.1
#> [91] GenomicAlignments_1.34.1 AnnotationDbi_1.60.2
#> [93] memoise_2.0.1 IRanges_2.32.0Conclusion
These benchmarks demonstrate that rgbio provides
substantial performance improvements for GenBank file parsing in R,
making it an excellent choice for both interactive analysis and
large-scale genomic data processing workflows. The Rust-powered backend
delivers the speed improvements while maintaining the familiar R
interface that researchers expect.
For the most up-to-date benchmarks and methodology, visit the rgbio GitHub repository.